在机器学习理论中,回归和分类是最经典的两个有监督学习问题,而聚类则是最经典的无监督学习方法。
其中,线性回归模型是最简单的一个回归模型,我们以此为例进行介绍。
线性回归的R语言实现
由于线性回归在传统的一元概率论与数理统计课程中已经介绍过了,所以这里直接介绍他的R语言实现。
线性回归的R语言实现,核心函数是lm。
而在概率论与数理统计的课程中应该也介绍过广义线性回归模型,例如$y=e^{ax+b}$次方,只需要两边取对数就又是线性回归了。
这种广义线性回归模型在R语言中借助函数glm实现。
例如:
|
|
在这里lm函数语法是
|
|
formula: 一个公式对象,指定了因变量和自变量之间的关系。例如,y ~ x表示y是因变量,x是自变量。data: 一个数据框,包含了模型中的变量。...: 其他参数,如method指定求解方法。
在本例中lm的summary是:
|
|
-
Call: 显示了用于拟合模型的
lm()函数的调用。 -
Residuals: 是每个数据拟合后的残差,可以看到他们接近于零,这说明模型完美地拟合了数据。
-
Coefficients是系数的意思:
-
(Intercept): 截距项。 -
x: 自变量x的系数。表中的第一列是各系数的估计值。
第二列是标准差。
第三列是进行显著性检验时需要的t统计量的值。
第四列是p-value
-
-
Residual standard error: 残差的标准误差,如果非常小,就说明模型完美地拟合了数据。
-
Multiple R-squared: 决定系数为 1,这意味着模型解释了所有变异。
-
F-statistic: F统计量非常大,p值非常小,这表明模型整体上是显著的。
而glm的summary内容和lm的是一样的。
需要注意的是,glm在调用的时候要额外给出family表示你要对数据进行怎样的变换。
R语言提供了很多family,例如
|
|
我无法逐一说明,感兴趣就自己上网查询。
在这里,如果选择poisson就是我们前面举例子的那个情况,拟合$y=e^{ax+b}$次方。
但是回归分析其实不能光看summary的报告,还得进行回归诊断。
需要通过残差分析验证拟合后的残差是否合理,通过影响分析观察数据有没有异常点或强影响点,通过共线性诊断判断变量是否有较强的共线性。
在R语言中进行残差分析只需要使用函数residuals(fit) rstandard(fit) rstudent(fit) (fit为函数lm()生成的对象)就可以计算模型的残差、标准化残差和学生化残差。
然后可以绘制这样的残差分析图:
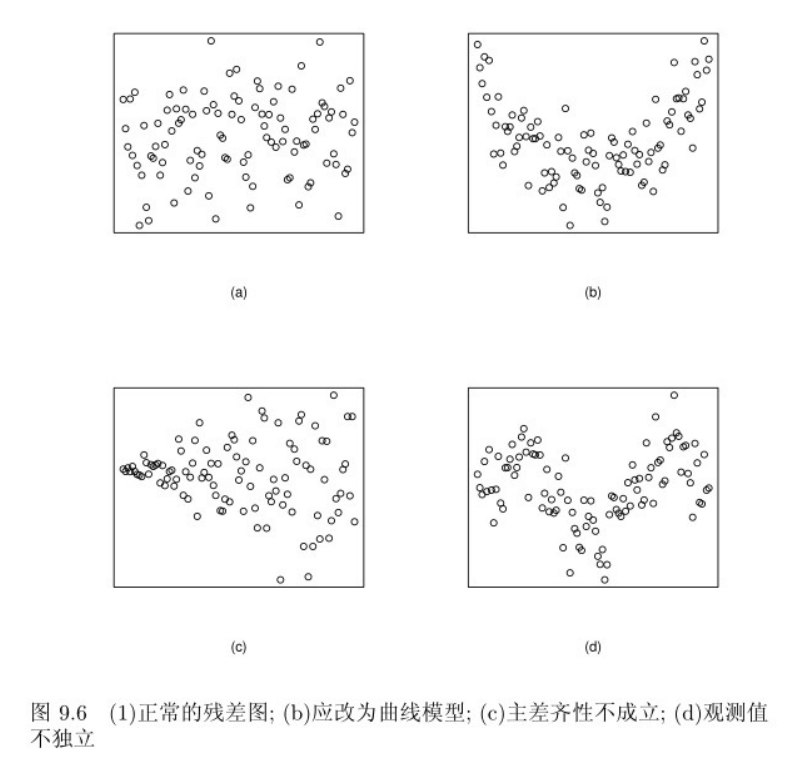
上图中只有图a是正常的残差,具有随机性。
b图是很明显的二次曲线,应该在拟合的时候引入交叉型或平方项。
图c残差在左边靠的很近,右边离得很远呈现喇叭口样式,我们说这种模型主差齐性不成立,你可以试着给Y取个对数、倒数、开平方或者进行box-cox变换再拟合看看。
图d的话就是观测值不独立。总之,只有图a才是最理想的残差图。
如果要进行影响分析,也就是查看数据有没有异常点或者强影响点,可以计算数据的标准化残差,如果某个数据的标准化残差大于2小于3,则他是可疑点。但如果他的标准化残差比3大,按照正态分布的$3\sigma$原则,他是异常点。
当然,其实还可以用帽子矩阵计算杠杆值,如果杠杆值比较大就说明那个样本是强影响点,或者计算库克距离(其实统计学里除了马氏距离还有绝对值距离、闵可夫斯基距离、切比雪夫距离、兰氏距离等很多距离),如果样本的库克距离大于1则是异常点。不过这两个比较进阶高级,你大概率没听过他的理论,也就不再介绍了。在R语言中通过函数influence.measures(fit模型)就可以直接计算前面说的什么库克距离啊之类的统计量了。
至于共线性诊断,主流的有3种方法,分布是特征值法、条件指数法、方差碰撞因子法。在此只介绍方差膨胀因子法。
在R语言中,安装扩展包DAAG,然后使用函数vif(fit模型,计算精确到小数点后几位)就可以计算方差膨胀因子了。
一般我们认为,如果方差碰撞因子>10则模型有很强的共线性问题,这时候需要用自回归模型来拟合而不是线性模型。
最后补充一嘴,线性回归还可以通过计算参数CP/AIC/BIC来衡量其优越程度,一般说来这三个参数越小越好。
线性判别分析LDA
在机器学习中有很多能实现分类问题的算法,如神经网络(好好好上来就开大是吧)、决策树、朴素贝叶斯分类器、支持向量机分类器SVC等等。我们这里就介绍最最简单的距离判别和线性判别分析lda。
所谓距离判别,简单地不能再简单了。
就是一个新样本离哪个总体近就认为这个样本属于哪个总体。
那什么是样本离总体的距离呢?
就是样本离该总体的均值的距离。
好,说完了。
距离判别只需要你选定一个距离,例如马氏距离。
然后你计算已知总体的均值。
接下来,每来一个新的样本,你就计算这个样本到各个总体的均值的距离,然后哪个总体到这个样本的距离最小,这个样本就分类为哪个样本。
线性判别分析LDA是一种fisher判别法,他先将数据进行线性变换,希望最大化两个或多个群体之间的距离。然后再进行判别。
也就是说,当你收到两个群体的数据的时候,你首先计算他们的均值$y_1,y_2$,然后施加线性变换得到$z_1=a^Ty_1,z_2=a^Ty_2$。
然后要找a,使得$z_1,z_2$的马氏距离最远。
而方差未知情形的两正态总体的方差我们在上一期已经说过,就是$(\frac{1}{n_1}+\frac{1}{n_2})a^TS_{pl}a$
因此我们要最大化的马氏距离的平方为$t^2(a)=\frac{(a^T(y_1-y_2))^T(a^T(y_1-y_2))}{(\frac{1}{n_1}+\frac{1}{n_2})a^TS_{pl}a}$
只需要使用柯西不等式$(a^Tb)^2\le(a^TWa)(b^TWb)$即可求解,得到$a=S_{pl}^{-1}(y_1-y_2)$。
其中a被称为判别函数系数,$z=a^Ty$则是fisher线性判别函数。
但是以上是两群体的情况,如果是多群体就麻烦多了。
这里只说最终结论。
需要构造组间离差矩阵$B=\sum_{k=1}^g(\overline y_k-\overline y)(\overline y_k-\overline y)^T$和组内离差矩阵$W=\sum_{k=1}^g\sum_{i=1}^{k_n}(y_{ki}-\overline y_k)(y_{ki}-\overline y_k)^T$
然后求矩阵$W^{-1}B$的特征值和特征向量。其中最大的特征值对应的特征向量就是a。
当求出a以后,就可以将所有的数据进行变换,从而得到一个一维的数据。
所以从某种意义上看,fisher判别分析也实现了一种降维。
而当数据变成这样的一维数据以后,我们再使用任意算法进行判别就会变得非常容易了,例如继续使用刚刚的距离判别法。
在R语言中只需要使用MASS库的lda函数就可以很简单地实现fisher线性判别分析了,例如:
|
|
等级聚类/层次聚类/系统聚类
接下来是聚类的话题。
聚类是一种无监督学习算法。
他和前面说的判别分析的不同之处在于,判别分析需要你先喂一些数据,告诉模型哪些数据属于A类,哪些属于B类,而无监督学习不需要。
这里要介绍的是一种最为简单的聚类方法,叫做层次聚类,也叫做系统聚类,还叫做等级聚类。
他的核心就是,让人来聚类啊哈哈哈。
说错了。
他的思想就是我每次都将距离最近的两个样本合并,认为他们属于同一个类别。
然后算法什么时候停止呢?等所有样本都被划分为一个类的时候停止,然后画出一张分析图,最后交由人工判别。
例如:
|
|
我们先用rnorm生成标准正态分布的随机数,把它放在10*10的矩阵里。
然后调用hclust函数进行层次聚类,最后进行绘图,你会收获这样的聚类图。
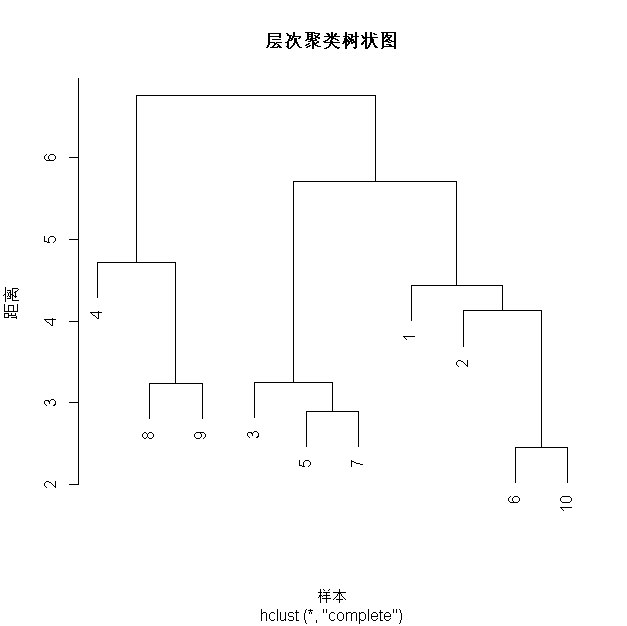
这个聚类图是从下往上读的,首先因为样本6和10距离最近,所以先将6和10合并为一个总体,此时得到右下角第一个合并,他的纵坐标是合并时的距离。
接着,5号和7号进行了合并,然后是3号和(5,7)这个整体进行了合并。
然后8号和9号合并,2号和(6,10)进行合并,1号和(2,6,10)进行合并。4号和(8,9)进行合并。
然后(3,5,7)与(1,2,6,10)进行合并。
最后(4,8,9)与(3,5,7,1,2,6,10)进行合并,到这里所有元素都归于一个类的,算法结束。
通过这张图,接下来由我们人工进行聚类。
例如我就想聚1类,那么就取最后一次合并的结果也就是1-10全部。
如果想分两类,就取倒数第二次合并的结果也就是(4,8,9)一类,其余一类。
想分三类就取倒数第三次的结果,以此类推。
借助函数cutree(hclust_result, k = 3)可以直接得到分3类的结果,这样你就不用自己手工去数了。
这就是层次聚类。
但是在层次聚类的时候,结果可能会因为细节的不同而不同。
例如我可能认为,所谓的距离是欧氏距离,也可能认为是马氏距离等等。
我可能认为,一个样本到总体的距离是指样本到总体的均值的距离,样本到总体最近(或最远)的点的距离等等。
我可能认为一个总体到另一个总体的距离是他们均值的距离,也可能认为是各个样本点距离的均值,等等。
反正就是有这么多变化。
k-means聚类
在R语言中实现k-means聚类非常简单。
|
|
而k-means本身也非常简单。
k-means和层次聚类不同,他需要用户先指定要分为k个类。
然后算法会随机选择k个初始聚类中心,接着将各个样本分类给距离他最近的距离中心。
然后再把各聚类中心的均值作为新的聚类中心。
依次重复,直到聚类中心不再移动。(收敛了)
就这么几句话就说完了,而且没有什么计算。